Amyloïdose cardiologie
Wild-type ATTR amyloïdose
Wanneer amyloïdfibrillen zich spontaan in weefsels en organen ophopen.
Epidemiologie | Symptomen | Diagnose | Behandeling
Patiënten met wildtype ATTR amyloïdose kunnen de ziekte niet doorgeven aan hun familie, in tegenstelling tot patiënten met de erfelijke vorm. Nog een verschil: erfelijke ATTR heeft meestal een duidelijke oorzaak, namelijk een gemuteerd gen dat codeert voor het transthyretine eiwit. Bij het verworven type is dat niet zo duidelijk; amyloïd lijkt zich ‘zomaar’ op te stapelen.1,2
Cardiale betrokkenheid is dé uiting van wildtype ATTR (ATTRwt). Een patiënt met cardiale amyloïdose heeft een hart waarvan de extracellulaire ruimte van het myocard is geïnfiltreerd door amyloïdefibrillen. Het gevolg? Cardiomyopathie met ernstige klachten en een sombere prognose. De meeste patiënten hebben niet lang meer te leven en overlijden door hartfalen of plotse hartdood.3

Gissen geblazen: de epidemiologie van wildtype ATTR
De incidentie en prevalentie van ATTRwt berekenen is nogal een uitdaging. De belangrijkste reden? Onderdiagnose.
Een patiënt kan bijvoorbeeld gediagnosticeerd zijn met (onbegrepen) hartfalen. Terwijl diegene eigenlijk ATTRwt als onderliggende aandoening heeft. Door zulke gemiste diagnoses bestaat het risico dat de cijfers onderschatten hoe vaak de ziekte voorkomt. Toch zijn er 2 studies gepubliceerd waarin gegevens over de prevalentie staan.
González-López et al. (2015) rapporteerden bijvoorbeeld dat ATTR amyloïdose een prevalentie heeft van 13% in een patiëntencohort met diastolisch hartfalen.4
En Castano et al. (2017) rapporteerden een prevalentie van 16% bij een patiëntengroep die een aortaklepvervanging onderging voor aortastenose.5 De geschatte prevalentiecijfers in deze studies hebben voornamelijk betrekking op patiënten met ATTRwt.
Hoe zit het dan met de genetisch overdraagbare vorm van ATTR? Daar kunt u meer over lezen op de pagina over erfelijke ATTR amyloïdose. Of kijk onderstaande animatie en ontdek in 3,5 minuut wat de belangrijkste verschillen tussen beide vormen zijn.
De symptomen van wildtype ATTR amyloïdose
ATTRwt leidt tot een progressieve toename in de dikte van het myocard; mogelijk zowel van de rechterventrikel als van de linkerventrikel. Hetzelfde geldt voor het interatriale septum en de atrioventriculaire kleppen.20
Het resultaat is hartfalen, vaak met behouden ejectiefractie (HFpEF). De meest voorkomende symptomen die daarbij horen, zijn:6
- vermoeidheid;
- dyspneu;
- oedeem.
Verder kan een patiënt met cardiale amyloïdose zich presenteren met symptomen zoals: aritmiën in de vorm van ventriculaire tachycardie, geleidingsstoornissen en pompfunctiestoornissen.6
Patiënten met ATTRwt kunnen ook een aantal andere klachten krijgen die niets met het hart te maken hebben. Want als amyloïdose systemisch is, dan kunnen de amyloïdfibrillen zich niet alleen ophopen in het hart, maar ook in andere organen en weefsels. Bij ATTRwt heeft bijvoorbeeld tot wel 50% van de patiënten een voorgeschiedenis van bilateraal carpaletunnel-syndroom.7
Bent u op zoek naar de oorzaak van hartfalen bij uw patiënt? Of heeft u het vermoeden dat er sprake is van ATTRwt als onderliggend lijden? Bekijk onderstaand schema met de belangrijkste aanwijzingen die in verband zijn gebracht met deze aandoening.

De diagnose van wildtype ATTR amyloïdose
U heeft verschillende diagnostische instrumenten tot uw beschikking om ATTRwt op te sporen. Bij voorkeur bestaat de diagnostiek uit meerdere van deze onderzoeken, zodat u de testuitslagen uiteindelijk kunt combineren voor een definitieve diagnose.2 De belangrijkste testen voor de diagnose van ATTRwt zijn:2
- echocardiografie en een ECG;
- geavanceerde beeldvormingstechnieken zoals een MRI-scan en botscintigrafie;
- laboratoriumonderzoek;
- genetische testen.
-
Het ECG
Beschrijving tonenEen bekend ECG-kenmerk van ATTRwt is een laag of normaal QRS-voltage, ondanks hypertrofie van het linkerventrikel.8
Hoe is dit te verklaren? Een verdikte wand is het gevolg van zich opstapelende amyloïdfibrillen, en niet van vergrootte hartspiercellen.8
Let op, bij cardiale amyloïdose is niet altijd sprake van de ‘klassieke’ lage QRS-voltages. Bij cardiale amyloïdose zien we een discrepantie tussen de toegenomen dikte van het myocard op een echo en de omvang van de voltages op het ECG. Het is daarom van belang om niet alleen naar lage voltages te zoeken. Waarom? Dit ECG-kenmerk is pas in een laat stadium van de ziekte waar te nemen. En dat maakt het wellicht minder nuttig om cardiale betrokkenheid snel te diagnosticeren.9
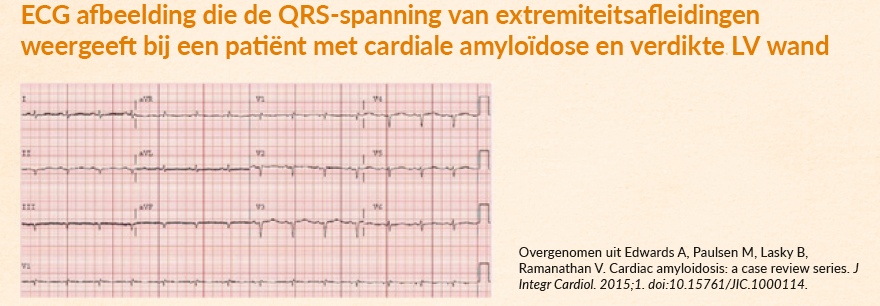
Figuur 1: Voorbeeld van ECG afbeelding die de QRS-spanning van extremiteitsafleidingen weergeeft bij een patiënt met cardiale amyloïdose en verdikte LV wand. -
De echo
Beschrijving tonenBij cardiale amyloïdose is extracellulaire infiltratie van amyloïdfibrillen verantwoordelijk voor een verdikte linkerventrikelwand (> 15 mm). Die wand is bij ATTR amyloïdose vaak dikker dan bij AL amyloïdose. Maar dat is niet altijd zo. De mate van verdikking is namelijk niet alleen afhankelijk van het type amyloïdose, maar ook van het stadium van de ziekte waarin de patiënt zich bevindt.
Dus, ziet u op de echo dat de wandikte van het hart van uw patiënt is toegenomen, terwijl daar geen duidelijke oorzaak voor is? Dan kan dat wijzen op cardiale amyloïdose.
Verminderde longitudinale strain is een andere aanwijzing op de echo die het vermoeden dat uw patiënt cardiale amyloïdose heeft, kan doen toenemen. Hierbij gaat het om een longitudinale strain die verminderd is in het basis- en middenwandsgebied. Terwijl de strain in de apex vaak wordt gespaard of behouden blijft wat resulteert in de kenmerkende ‘kers op de taart’, zoals u in onderstaand figuur ziet. 8,10,11
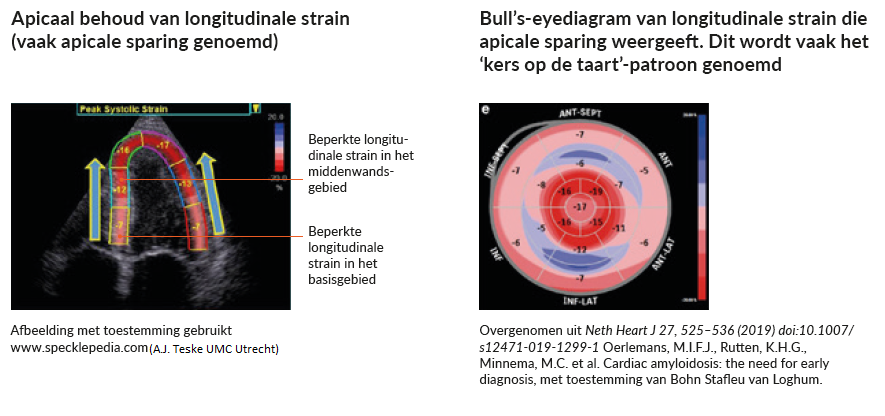
Figuur 2: Voorbeelden van ECHO strain imaging die apicaal behoud weergeven.
-
De MRI
Beschrijving tonenOok een MRI kan u specifieke kenmerken voor cardiale amyloïdose tonen, bijvoorbeeld:
- transmurale of subendocardiale late gadolinium enhancement beeldvorming (LGE);
- diffuus atriaal LGE;
- rechterventrikel LGE;
- suboptimale nulstelling als gevolg van veranderde gadoliniumkinetiek;
- verhoogd extracellulair volume en verhoogde native T1-waardes.8
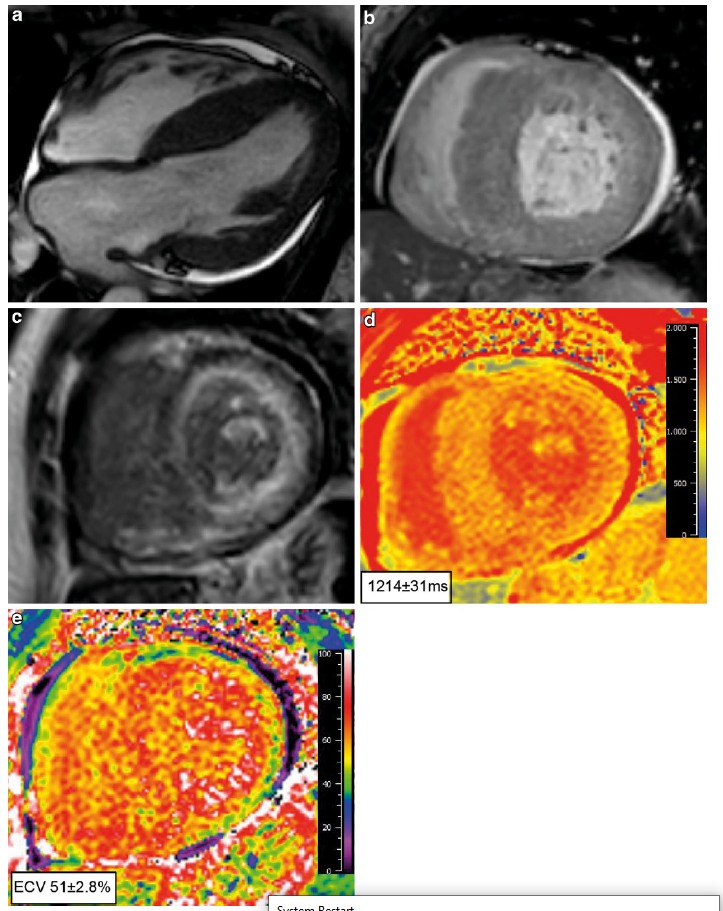
Een 4-kamer en korte as beeld met linkerventrikelhypertrofie, voornamelijk in de septale regio (a,b). Corresponderend korte as beeld dat subendocardiale LGE laat zien. Let op het verminderde signaal van de bloedpoel (donker bloed), specifiek voor cardiale amyloïdose (c). Native T1-analyse met verhoogde T1-waardes (d) en een verhoogd extracellulair volume (e). Overgenomen uit Neth Heart J 27, 525 -536 (2019)doi:10.1007/ s12471-019-1299-1 Oerlemans, M.I.F.J., Rutt en, K.H.G., Minnema, M.C. et al. Cardiac amyloidosis: the need for early diagnosis. Met toestemming van Bohn Stafl eu van Loghum. -
Het labonderzoek
Beschrijving tonenVerder is bloedonderzoek in het laboratorium noodzakelijk. Op basis van dat resultaat kunt u de diagnose AL amyloïdose snel bevestigen of verwerpen. De laborant onderzoekt serum en urine van de patiënt op de aanwezigheid van monoklonaal eiwit. Als u twijfelt over de betekenis van de uitslag, bijvoorbeeld als één van de drie labwaarden positief is, dan is overleg met de hematoloog aan te raden.8
Daarnaast zijn verhoogde waarden van NT-proBNP en troponine aanwijzingen voor cardiale betrokkenheid.12,13 Ten eerste is het NT-proBNP niveau vaak onevenredig verhoogd. Ten tweede is troponine chronisch verhoogd zonder stijging of daling. Beide bepalingen hebben een prognostische betekenis.16,17
-
De botscan
Beschrijving tonenBotscintigrafie is een niet-invasieve manier om de diagnose ATTR amyloïdose te bevestigen, in tegenstelling tot een hartbiopt. Bovendien is het een zeer gevoelige diagnostische beeldvormingstechniek waarmee u de verschillende vormen van amyloïdose van elkaar kunt onderscheiden.14,15
Er zijn verschillende radiotracers die een sterke affiniteit met amyloïd hebben en dus geschikt zijn voor de diagnose van amyloïdose. Bijvoorbeeld: 99mTc-PYP, 99mTc-DPD en 99mTc-H MDP.16
De radiotracers zijn vooral gevoelig en specifiek voor amyloïdfibrillen die het hart hebben geïnfiltreerd. Cardiale lokalisatie van een radiotracer is mogelijk bij ATTR amyloïdose en, in mindere mate, bij AL amyloïdose (zoals graad 1 traceropname, zie hieronder).14,15
Wat is nodig om de diffuse opname van de radiotracer in het myocard te bevestigen? In ieder geval een evaluatie van de planar- en de SPECT-beelden, als onderdeel van de visuele interpretatie van de botscan.16
Als de opname van de radiotracer te zien is op de SPECT-beelden, dan kunt u de graad van de tracerstapeling beoordelen met semikwantitatieve sorteringen. En die traceerstapeling vergelijkt u uiteindelijk met de traceropname in de ribben (zie onderstaande figuur), volgens graad 0 tot en met 3:16
- Graad 0: geen cardiale opname van de tracer en normale opname in de ribben.
- Graad 1: traceropname in het myocard, maar minder dan in de ribben.
- Graad 2: opname in het myocard is gelijk aan de traceropname in de ribben.
- Graad 3: traceropname in het myocard is groter dan opname in de ribben.
In onderstaande figuur ziet u de visuele vergelijking, zowel op planar- als SPECT-beelden.
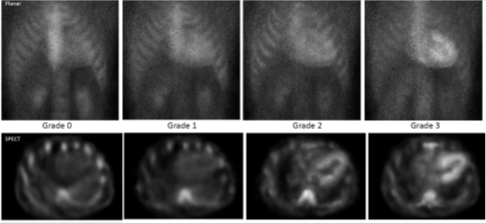
Visuele vergelijking van tracer opname in het myocard en de ribben, zowel planar als SPECT.16 Is de botscintigrafie negatief of onduidelijk? En is er reden om de patiënt te blijven verdenken van amyloïdose? Overweeg dan een hartbiopt met congoroodkleuring, immunohistochemie en/of massaspectometrie.16
Wilt u meer informatie over medische beeldvorming? Download de samenvatting van de ‘Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis (MECR)' via onderstaande link.
De behandeling van wildtype ATTR amyloïdose
Meestal wordt de behandeling van ATTRwt op 2 manieren aangepakt. Dat gebeurt tegelijkertijd. De ene manier richt zich op congestief hartfalen dat kan optreden als gevolg van cardiale betrokkenheid. De andere manier is bedoeld om het ziekteproces van amyloïdose te vertragen door verder afzetting van amyloïdfibrillen te voorkomen.20
Hartfalentherapie18,21,22
De basis van therapie voor hartfalen bestaat uit een verminderde zoutinname en farmacotherapie met diuretica en aldosteronantagonisten. Maar geen bètablokkers.
Waarom?
Die kunnen de hypotensie verergeren en de myocardiale contractiliteit verlagen, omdat ze een negatief inotroop effect hebben. Ook digoxine en calciumantagonisten zijn gecontra-indiceerd. Implanteerbare cardio-defibrillatoren en pacemakers zijn daarentegen wel geïndiceerd.
Het ziekteproces van amyloïdose vertragen
Voor deze aandoening is de behandeling gericht op het transthyretine-eiwit (TTR) dat zich verkeerd vouwt en opstapelt. Dat gebeurt met een transthyretine stabilisator.23
Deze stabilisator bindt zich selectief en met hoge affiniteit aan TTR (een tetrameer). Daardoor dissocieert het minder snel in monomeren. En dat draagt bij aan de stabiliteit van het eiwit. Die dissociatie is de snelheidsbepalende stap van het proces dat leidt tot de vorming van amyloïd. Dus daar ingrijpen vormt de rationale voor het gebruik van stabilisators bij patiënten met ATTR amyloïdose.23
Wilt u meer weten over de behandeling van amyloïdose? Ga naar de website van het Expertisecentrum Amyloïdose (onderdeel van het UMCG). Of neem direct contact op met dit expertisecentrum of dat van het UMC Utrecht.
Bronnen
1. Donnelly J, et al. Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 Suppl 3):12–26
2. Siddiqi O, et al. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28:10–21
3. Connors L, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016 Jan; 19;133(3):282-90
4. González-López E, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–94
5. Castano A, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–87
6. Rapezzi C, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-124
7. Nakagawa M, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58-63
8. Oerlemans M, et al. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27, 525–536
9. Cyrille N, et al. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114(7):1089-1093
10. Narotsky D, et al. Wild-type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol. 2016;32(9):1166.e1-1166.e10
11. Rapezzi C, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-1212
12. Kumar S, at. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–95.
13. Gillmore J, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39:2799–806.
14. Van den Wyngaert T, et al. The EANM practice guidelines for bone scintigraphy. Eur J Nucl Med Mol Imaging. 2016;43:1723–38
15. Gillmore J, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12
16. Dorbala S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. doi:10.1007 /s12350-019-01760-6
17. Maurer M, et al. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis Circulation. 2017;135:1357–77
18. Castaño A, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178
19. Sperry BW, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040-2050
20. Fikrle M, et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa. 2013;55(1):e60-e75
21. Dubrey SW, et al. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97:75-84
22. Nativi-Nicolau J, et al. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018 Sep;33(5):571-579
23. Johnson SM, et al. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012 Aug 10;421(2-3):185-203
Ook interessant
-
Neurologische uiting van ATTR amyloïdose
Gezondheidswinst door een snelle diagnose
Differentiaaldiagnose | Klinische presentatie | Diagnose | Behandeling
In het begin sluimeren de symptomen. Pas na gemiddeld 4 jaar kondigt de neurologische vorm van ATTR amyloïdose (ATTR-PN) zich aan: de patiënten krijgen symptomen van polyneuropathie. Vanaf dat moment is de levensverwachting, zonder behandeling, gemiddeld 6 tot 12 jaar.1
-
Talkshow 3: ATTR & erfelijkheid
In gesprek met Paul van der Zwaag (klinisch geneticus, UMCG) en Hans Nienhuis (klinisch immunoloog/internist, UMCG).